\[
\delta_{plot} \sim \mathcal N (\phi_{BA,plot} \times \gamma_{site}, \sigma_{\delta})
\]
Fit
Code
data <- read_tsv("../modelling/data/derived_data/data_rec.tsv", col_types = cols()) %>%
select(-y) %>%
filter(variable %in% c("ba", "diversity_q1_gen")) %>%
select(-sitenum, -plotnum) %>%
pivot_wider(names_from = variable, values_from = value) %>%
na.omit() %>%
filter(!(site %in% c("Manare", "Montagne Tortue", "Nelliyampathy",
"Uppangala", "BAFOG", "Sao Nicolau",
"Kabo", "Mil", "Corinto",
"Peixoto", "Iwokrama", "Antimary", "Peteco"))) %>%
mutate(sitenum = as.numeric(as.factor(site))) %>%
mutate(plotnum = as.numeric(as.factor(paste(site, plot)))) %>%
arrange(sitenum, plotnum, rel_year) %>%
gather(variable, y, -plot, -year, -site, -treatment, -harvest_year,
-longitude, -harvest_year_min, -rel_year, -sitenum, -plotnum)
trajectories <- read_tsv("chains/joint_nointercept-diversity_q1_gen-trajectories.tsv")
parameters <- read_tsv("chains/joint_nointercept-diversity_q1_gen-parameters.tsv")
parameters
# A tibble: 709 × 14
variable mean median sd mad q5 q95 rhat ess_bulk
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 lp__ 16726. 1.67e+4 1.25e+2 1.66e+2 1.65e+4 1.69e+4 1.89 5.69
2 gamma 0.0292 3.48e-2 4.03e-2 3.97e-2 -4.18e-2 8.85e-2 1.46 7.83
3 gamma 0.0399 3.06e-2 3.37e-2 2.73e-2 -2.49e-3 1.07e-1 1.50 7.41
4 gamma 0.0190 2.39e-2 3.33e-2 3.22e-2 -4.23e-2 6.69e-2 1.42 8.25
5 gamma -0.00159 -3.26e-4 4.49e-2 4.27e-2 -7.72e-2 7.01e-2 1.04 74.0
6 gamma 0.237 2.39e-1 1.08e-1 1.05e-1 5.64e-2 4.08e-1 1.00 1851.
7 gamma 0.00971 9.03e-2 1.91e-1 7.31e-2 -3.92e-1 1.88e-1 1.53 7.17
8 gamma 0.127 1.24e-1 3.90e-2 3.81e-2 6.55e-2 1.93e-1 1.18 15.4
9 gamma 0.00607 3.46e-2 6.65e-2 2.86e-2 -1.22e-1 6.96e-2 1.53 7.17
10 gamma -0.0134 -1.24e-2 3.58e-2 3.48e-2 -7.37e-2 4.36e-2 1.14 18.9
# ℹ 699 more rows
# ℹ 5 more variables: ess_tail <dbl>, sitenum <dbl>, site <chr>, plotnum <dbl>,
# plot <chr>
IDH - H2
Code
area <- data %>%
filter(variable == "area") %>%
select(site, plot, y) %>%
unique() %>%
group_by(site) %>%
summarise(area = mean(y))
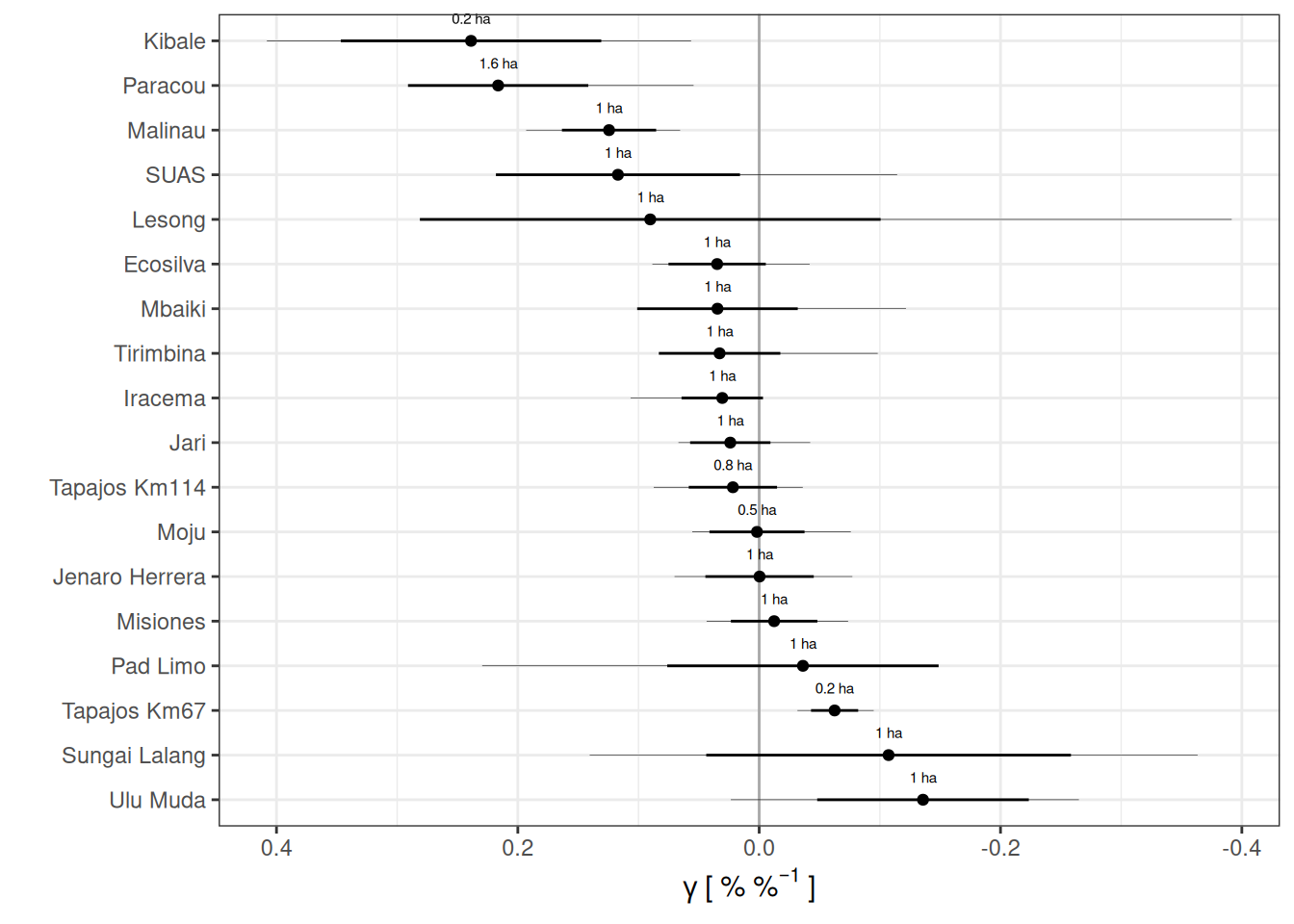
filter(parameters, variable == "gamma") %>%
left_join(area) %>%
ggplot(aes(median, reorder(site, median))) +
geom_vline(xintercept = 0, col = "darkgrey") +
geom_segment(aes(x = (median - sd),
xend = (median + sd))) +
geom_segment(aes(x = q5, xend = q95), size = .1) +
geom_point() +
geom_text(aes(label = paste(round(area, 1), "ha")), nudge_y = .5, size = 2) +
theme_bw() +
scale_x_reverse(expression(gamma~"["~"%"~"%"^{-1}~"]")) +
ylab("") +
scale_color_viridis_c()
Code
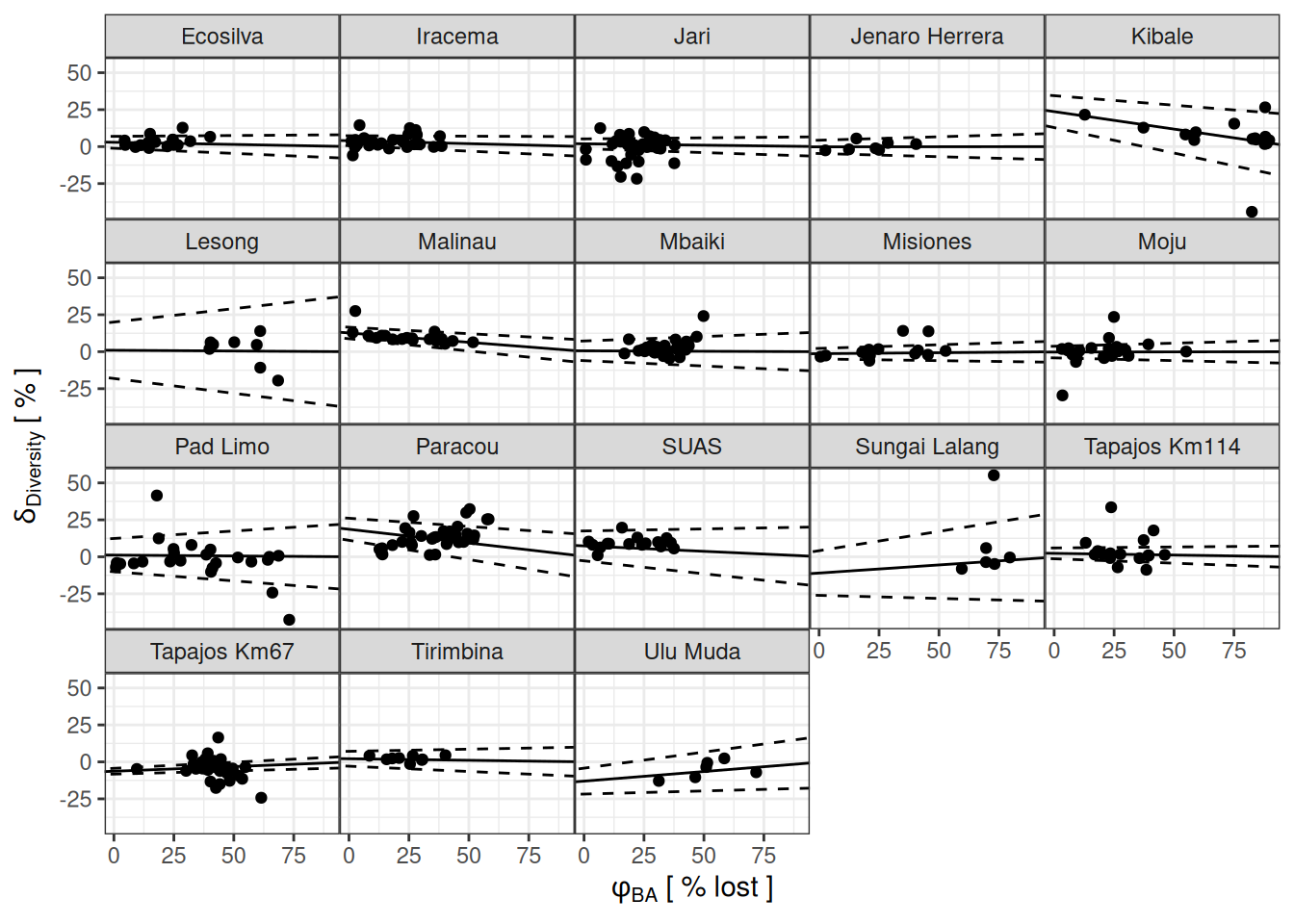
pars_s <- filter(parameters, variable == "gamma")
filter(parameters, variable %in% c("delta", "phiba")) %>%
select(site, plot, variable, median) %>%
pivot_wider(names_from = variable, values_from = median) %>%
ggplot(aes((1-phiba), delta)) +
facet_wrap(~ site) +
geom_abline(aes(slope = -mean,
intercept = mean, group = site),
data = pars_s) +
geom_abline(aes(slope = -mean-sd,
intercept = mean-sd, group = site),
data = pars_s, linetype = "dashed") +
geom_abline(aes(slope = -mean+sd,
intercept = mean+sd, group = site),
data = pars_s, linetype = "dashed") +
geom_point() +
theme_bw() +
xlab(expression(phi[BA] ~ "[ % lost ]")) +
ylab(expression(delta[Diversity] ~ "[ % ]")) +
theme(panel.spacing = unit(0, "lines")) +
scale_x_continuous(labels = ~.*100) +
scale_y_continuous(labels = ~.*100)
Comparison
Code
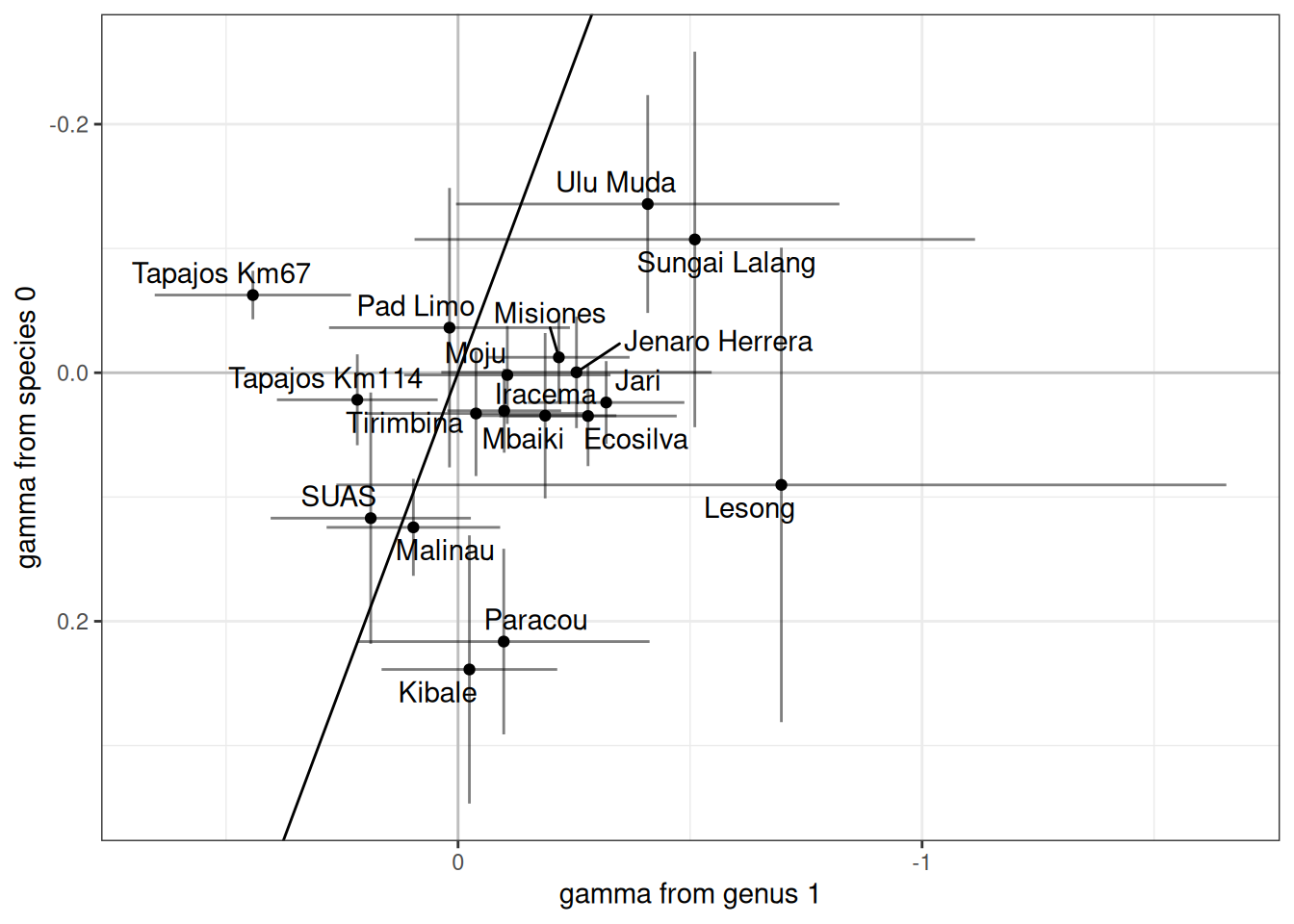
read_tsv("chains/joint-diversity_q1_gen-parameters.tsv") %>%
filter(variable == "gamma") %>%
left_join(filter(parameters, variable == "gamma") %>%
select(site, median, sd, q5, q95) %>%
rename(median_new = median, sd_new = sd,
q5_new = q5, q95_new = q95)
) %>%
filter(!is.na(median_new)) %>%
ggplot(aes(median, median_new)) +
geom_hline(yintercept = 0, col = "grey") +
geom_vline(xintercept = 0, col = "grey") +
geom_point() +
geom_segment(aes(x = median - sd, xend = median + sd), alpha = .5) +
geom_segment(aes(y = median_new - sd_new, yend = median_new + sd_new), alpha = .5) +
ggrepel::geom_text_repel(aes(label = site)) +
theme_bw() +
geom_abline() +
xlab("gamma from genus 1") +
ylab("gamma from species 0") +
scale_x_reverse() +
scale_y_reverse()